x



Multi-genome assemblies called pangenomes can capture genetic diversity in a species, but researchers are still working out how best to build and explore them.
Long-read systems make it easier to assign a given sequence to a chromosome copy.Credit: SCIEPRO/Getty
The word ‘reference’ conveys authority, signifying a trusted resource against which new information can confidently be assessed. That was true of encyclopaedias and atlases, and it’s true of reference genomes — ultra-high-accuracy maps that describe the complete sequence of a species’ chromosomal DNA.
But it’s an open secret that individual reference genomes do a poor job of providing real-world biological blueprints. David Edwards, a bioinformatician at the University of Western Australia in Perth, recalls a colleague who wanted to study gene expression in wheat variants using a single, well-studied strain. “We’ve shown that there’s like 20,000 genes that are in commercial wheat lines but are not in that reference,” he says. “You’re missing a huge amount unless you take account of that.”
With highly complex and variable genomes, plants are an extreme example, but hidden diversity is everywhere. One comparison of 64 genomes of human individuals revealed nearly 16 million single-nucleotide differences and more than 2 million structural variants in which sequences were deleted or inserted1. This makes it impossible to define any one genome as ‘the reference’ against which all others can be compared. And, given that most genomes sequenced so far are from people of western European descent, key genomic insights for individuals of other backgrounds could be missed. “It’s kind of a nightmare to envision a genomic medicine practice that would work better for people of some ancestries and worse for people of other ancestries,” says Tobias Marschall, a computational-genomics researcher at Heinrich Heine University in Düsseldorf, Germany, and a lead author of that comparison study.
The solution is the pangenome: a composite reference, made from multiple genomes, that captures a wider range of variability and diversity at any given chromosomal site. Already an established tool for microbes and plants, pangenomes are finally reaching the vertebrate world. In July 2022, the Human Pangenome Reference Consortium (HPRC) published a preprint of a draft pangenome based on 47 individuals who represent a wide swathe of ethnic and geographic diversity2. Hundreds more genomes are now slated for incorporation into this assembly.
But pangenomes are still new enough that the field is grappling with how to package and explore them — and to persuade researchers to discard the familiar linear references of conventional genomics. “This is something that will take the whole field about a decade to transition,” predicts Benedict Paten, a computational-genomics researcher at the University of California, Santa Cruz (UCSC), and part of the HPRC. “You’ve got to demonstrate that it actually improves things for people — otherwise, what’s the point?”
As with so many genetics advances, the earliest demonstrations of pangenomics came from single-cell microbes. In 2005, a team led by Claire Fraser at the Institute for Genomic Research in Rockville, Maryland, and Rino Rappuoli at Chiron Vaccines in Siena, Italy, created an assembly of genomes from eight isolates of Streptococcus agalactiae, a bacterium responsible for potentially lethal infections in young children3. Each added genome brought dozens of new genes into the assembly — which they called a ‘pan-genome’ (‘pan’ being Greek for ‘whole’) — starkly highlighting the shortcomings of conventional references.

Closing in on a complete human genome
Microbial pangenomics is now a thriving area of research. Bernhard Palsson, a systems biologist at the University of California, San Diego, says that by 2013, his team had compiled 55 different strains of Escherichia coli into one pangenome4. By assessing how variants across genomes correlate with biological functions in these bacteria, they were able to link differences in metabolism and virulence to specific genes and chromosomal features. Since then, Palsson’s team has pushed the pangenome concept beyond the strain and species level to survey even more distantly related organisms, including a family of bacteria known as Lactobacillaceae. “We had 3,500 genomes or so to work with,” he says.
The first eukaryotic pangenomes came from the plant world, starting with the assembly, in 2014, of seven soya-bean genomes by a group led by crop geneticist Lijuan Qiu at the Chinese Academy of Agricultural Sciences in Beijing5. Crucial crops such as wheat, maize (corn) and rice followed. “Most of the major species now have pangenomes,” says Jacqueline Batley, a plant-genomics researcher at the University of Western Australia and a close collaborator of Edwards’s. Plant biologists are using these resources to develop improved variants that incorporate genetic features associated with hardiness against drought or pathogens, increased yield and other valuable traits.
Progress in the human pangenome realm has been propelled by innovations in sequencing and genome assembly that allowed a network of researchers across the globe to publish the first truly complete ‘telomere-to-telomere’ genome sequence6 in March 2022. Karen Miga, the UCSC geneticist who co-led this effort, says the completion in 2019 of the first full human X chromosome sequence — with its messy assortment of highly repetitive elements — was like “shooting a flare gun up into the air”, signalling that the community finally had the capacity to pursue a human pangenome reference. “It was just a matter of figuring out the right data-production and assembly strategy,” she says. The HPRC — for which Miga is a programme director — was launched that same year.
For the first wave of pangenomes, DNA sequences were largely collected using ‘short-read’ systems developed by biotechnology firm Illumina, based in San Diego, California, which are highly accurate but produce reads that are only about 100–200 nucleotides long. Researchers can assemble these fragments into ‘contigs’ that reveal relatively small differences such as single-nucleotide variants and ‘indels’ — insertions or deletions of a handful of nucleotides — but that cannot resolve larger structural variations. For this reason, early pangenomes typically mapped short-read-derived contigs from each specimen to an existing reference. This approach tends to produce gene-centric pangenomes that miss complex structural variation in individual genomes, which can play an important part in gene regulation and contain essential information about genome evolution.

Long road to long-read assembly
Still, these ‘map-to-pangenome’ approaches can be useful. Edwards and Batley say that their first attempt at a wheat pangenome based on short-read analysis, in 2017, was highly effective for determining which genes are absent or present in particular cultivars7. But this approach also undermines the whole purpose of creating a reference, by introducing biases on the basis of which genome serves as the pangenome’s foundation, such that one assembly could differ considerably from another.
A better solution is to build multiple reference-quality genomes and align those in an unbiased fashion, charting where they match and how they differ — an approach made feasible by the rapid evolution of ‘long-read’ sequencing technologies.
Longer reads have also simplified a second, thorny challenge. Humans — and many plant and animal species — are diploid, meaning that they carry two copies of every non-sex chromosome. Each copy has its own pattern of variations, known as a haplotype. Some species have more than two copies; wheat, for instance, contains six. This presents a baffling problem for short-read sequencing — how to assign a given read to a specific chromosome copy. “It’s like putting two giant puzzles together, and the pieces are so similar, you don’t know which one it goes to,” says Erich Jarvis, a neurogenetics researcher at the Rockefeller University in New York City. This, he adds, represents “one of the biggest problems to getting accurate genome assemblies”.
For the HPRC’s ‘first draft’ pangenome, Jarvis, Miga and their colleagues tackled the haplotype problem by using genome data from each DNA donor’s parents, giving insight into which sets of variants came from the mother and which from the father8. Long-read sequencing was essential here, because it allowed HPRC scientists to traverse sufficiently vast stretches of DNA to distinguish one chromosome from the other. By feeding the data from all three genomes into a software tool called Hifiasm, the researchers were able to recover diploid genomes for which each chromosome’s haplotype was effectively resolved, or ‘phased’.
Still, the 47 diploid genomes in this initial pangenome are not complete assemblies like the telomere-to-telomere genome. That effort exploited an unusual cell line in which both chromosome copies are identical. In true diploid cells, Jarvis says, the HPRC workflow typically yields not a single chromosome but hundreds of massive contigs, with gaps occurring in arrays of ultra-similar duplicated genes as well as the gnarly and repetitive centromeric regions that connect each chromosome’s gene-laden arms. The consortium is still struggling with how best to handle these problematic regions, he says.
Seven technologies to watch in 2022
The good news is that the current process covers the majority of the genome and can be largely automated. Marschall highlights Verkko, software developed by his former student Mikko Rautiainen while he was a postdoc at the US National Human Genome Research Institute in Bethesda, Maryland, that greatly simplifies diploid assembly. “Some chromosomes come back just in a single, fully phased contig,” he says. That should help the HPRC to meet its goal of assembling 350 diverse genomes for the first-generation human pangenome by 2024.
Consortium scientists have also identified experimental methods that allow them to physically link together sequencing reads that originate from the same chromosome — even over very long distances — eliminating the burdensome requirement of collecting and sequencing parental DNA. “I think now we are at a point where we can almost get telomere-to-telomere [assemblies] in the diploid setting with single samples,” says Marschall.
This leaves the essential question of how to depict a pangenome. The linear maps used to illustrate reference genomes over the past 20 years don’t work for assemblies comprising tens, hundreds or even thousands of individual genomes.
Most researchers in the field have converged on graph pangenomes as the best current solution to this problem (see ‘Visualizing a Pangenome’). These elaborate network diagrams collapse shared regions of genome sequence to the familiar flat line, but loop out into divergent paths at sites where variability can occur. Think of a city public-transport map, which presents default routes for trains. Maintenance, accidents or rush-hour schedules can cause trains to reroute on to other lines or skip stations, but there are limits on the number of detours. A graph-style map of the train line would capture both the invariant parts of a route and all detours that have been known to occur — essentially, describing the range of possible haplotypes for that line.
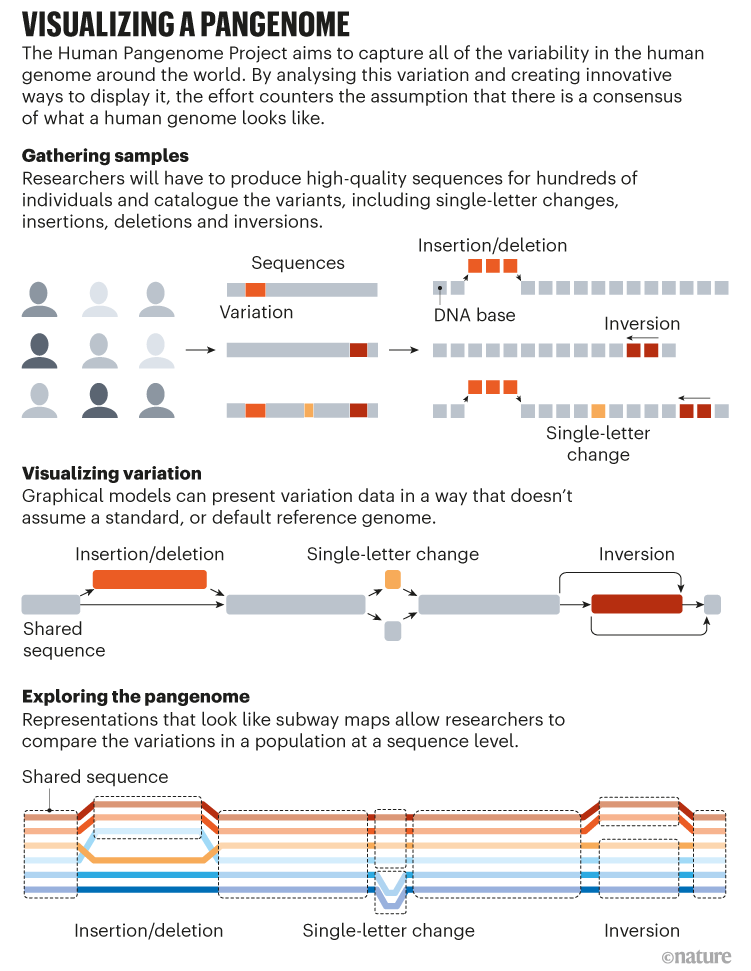
Computational-genomics researchers are still working out how best to build such graphs, and the HPRC’s draft-pangenome preprint explores several possibilities. One involves iterative assembly of individual diploid genomes, but although that approach could handle large structural variations nicely, “it didn’t bring base-level resolution”, Miga says. The other, more computationally intensive approach involves aligning all genomes simultaneously, which works well for gene-containing regions but struggles in repetitive, low-complexity chromosomal areas. “That’s why, intentionally, this paper has ‘draft’ in the title, to convey that it’s our first shot,” says Marschall.
Researchers building non-human pangenomes face steeper challenges. Edwards and Batley have found that human-centric graph-assembly software packages don’t work so well with plants, for instance. “We need some more tools,” says Edwards, noting that the greater complexity of plant genomes relative to human ones represents a crucial stumbling block. And Jarvis, who is also coordinating the Vertebrate Genomes Project, an initiative to build reference sequences for every vertebrate species on Earth, says the HPRC’s pipelines translate poorly to many of our animal relatives. “Even for this human pangenome, we’re finding that the assembly tools need to be tweaked a little bit more for different people,” says Jarvis.
There is also the challenge of getting the broader community on board. Past iterations of the human reference genome have been slow to percolate into general usage, and many clinical laboratories still have not adopted the current state-of-the-art reference, GRCh38. Plus, researchers outside the field might find this new reference format off-putting. “People are daunted by the graphs,” says Batley.
One solution is to build tools that keep the graph itself ‘under the hood’, and let researchers interrogate specific regions of the genome with more user-friendly graphical interfaces. Miga champions the idea of linking the human pangenome to GRCh38 sequence coordinates so that users of the current reference do not need to fully overhaul their analytical workflows. But promoting graph pangenome uptake will be a top priority for the HPRC in the coming year, she adds.
Ultimately, the best advertisement for pangenomes will be proof of their power, and pioneers in the field are enthusiastic about the secrets that a well-assembled multi-genome reference can unlock.

Again, microbial pangenomics is leading the way. Palsson points to a 2018 analysis of haplotype-specific traits that his team conducted using a pangenome comprising nearly 1,600 isolates of Mycobacterium tuberculosis9, the bacterium that causes tuberculosis. “We could associate that [genomic variability] with metabolic properties and elucidate antimicrobial-resistance mechanisms,” he says.
Similarly, plant pangenomes are helping researchers to home in on previously overlooked genes that confer a survival edge in harsh conditions. Zhixi Tian, a plant genomicist at the Chinese Academy of Sciences in Beijing, notes that many of these features reside in structurally variable regions that were absent from earlier reference genomes. “Usually for the stress-related traits, the genes that control them are duplicated,” says Tian. “The dosage difference makes the trait difference.”
Pangenome maps could prove equally powerful for uncovering the hidden variation that underlies complex developmental and medical conditions in humans. For example, the Paten group’s Giraffe algorithm can analyse millions of tiny snippets of the short-read sequencing data that is typically collected in clinical genomics and extrapolate which haplotype ‘route’ someone’s sequence follows through the graph, filling in the blanks about the rest of their genome. Jarvis also cites the possibility of creating focused pangenomes for medical and developmental conditions, such as autism spectrum disorder, and then comparing those against the baseline pangenome to identify divergent genomic features.
Another exciting possibility is integrating pangenome references with other biological information to provide a more holistic view of how chromosomal variation informs cellular function. For example, some researchers are creating ‘pantranscriptomic’ data sets that complement genomic data with RNA sequencing to study how DNA variation influences the quantity and structure of the resulting gene transcripts. And the HPRC team is collecting epigenetic data from its donor genomes to better understand the molecular-scale differences in gene expression between individuals.
“It’s not about just the base pairs,” Miga emphasizes. “We need to start building that type of annotation map on top of the pangenome, so it becomes a one-stop shop.”

